Health Advice
/Health

False offers of cash subsidies used to 'capture' health insurance customers, lawsuit alleges
A health insurance operation based in Broward County, Florida, used internet ads that falsely promised cash subsidies to sign up clients across the country and replace their agents, a lawsuit contends.
The scheme was carried out by Enhance Health LLC, TrueCoverage LLC, Speridian Technologies LLC, Number One Prospecting LLC and two individuals ...Read more
Atrium Health shared patient data with Facebook, class-action lawsuit alleges
A class-action lawsuit filed in North Carolina accuses Atrium Health of allowing Facebook and Google to access patient information online to use in targeted ads.
The plaintiffs, identified only as North Carolina-resident J.S. and Michigan-citizen J.R., allege they received spam mail and Facebook ads related to their medical conditions after ...Read more
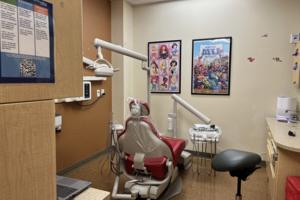
Doctors take on dental duties to reach low-income and uninsured patients
DENVER — Pediatrician Patricia Braun and her team saw roughly 100 children at a community health clinic on a recent Monday. They gave flu shots and treatments for illnesses like ear infections. But Braun also did something most primary care doctors don’t. She peered inside mouths searching for cavities or she brushed fluoride varnish on ...Read more

Mayo Clinic virologist offers perspective on avian influenza outbreak
Health officials around the globe, including the Centers for Disease Control and Prevention and the European Centre for Disease Control and Prevention, are monitoring the ongoing avian influenza outbreak. Also known as bird flu, the highly contagious viral disease typically spreads among birds, but can also infect livestock and, in rare cases, ...Read more
Ten doctors on FDA panel reviewing Abbott heart device had financial ties with company
When the FDA recently convened a committee of advisers to assess a cardiac device made by Abbott, the agency didn’t disclose that most of them had received payments from the company or conducted research it had funded — information readily available in a federal database.
One member of the FDA advisory committee was linked to hundreds of ...Read more

City-country mortality gap widens amid persistent holes in rural health care access
In Matthew Roach’s two years as vital statistics manager for the Arizona Department of Health Services, and 10 years previously in its epidemiology program, he has witnessed a trend in mortality rates that has rural health experts worried.
As Roach tracked the health of Arizona residents, the gap between mortality rates of people living in ...Read more

U.S. Supreme Court allows Idaho's ban on gender-affirming care to go into effect
The U.S. Supreme Court has ruled that Idaho’s ban on gender-affirming care for transgender minors can go into effect, overruling a lower court while the long-term constitutionality of the state’s law is still being litigated.
Idaho Attorney General Raúl Labrador asked the high court to allow the ban to go into effect in February, after a ...Read more

U.S. Supreme Court allows Idaho's ban on gender-affirming care to go into effect
The U.S. Supreme Court has ruled that Idaho’s ban on gender-affirming care for transgender minors may go into effect, overruling a lower court while the long-term constitutionality of the state’s law is still being litigated.
Idaho Attorney General Raul Labrador asked the high court to allow the ban to go into effect in February, after a ...Read more
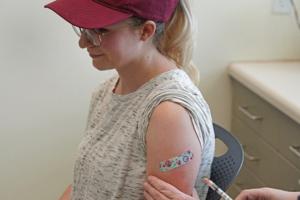
Why some adults may need another dose of measles vaccine
A rising number of measles cases in the U.S. this year is raising concerns over a comeback of a disease that was declared eliminated in this country 24 years ago.
A measles report from the Centers for Disease Control and Prevention last week concluded that the spike in cases means more public health efforts are needed to increase routine ...Read more

More kids are dying of drug overdoses. Could pediatricians do more to help?
A 17-year-old boy with shaggy blond hair stepped onto the scale at Tri-River Family Health Center in Uxbridge, Massachusetts.
After he was weighed, he headed for an exam room decorated with decals of planets and cartoon characters. A nurse checked his blood pressure. A pediatrician asked about school, home life, and his friendships.
This ...Read more

Colorado has lost dozens of autism clinics as state struggles to shore up funding
For the first time in years, Jay Ortengren has seen his 16-year-old son, Ethan, reach milestone after milestone as he lives with a severe form of autism.
Ortengren and his family uprooted their lives in search of the best treatment for Ethan after he was diagnosed as a young child. Finally, they seemed to find it when they moved to Jefferson ...Read more

To close racial gap in maternal health, some states take aim at 'implicit bias'
Countless times, Kenda Sutton-El, a Virginia doula, has witnessed her Black pregnant clients being dismissed or ignored by clinicians.
One woman was told by doctors that swelling, pain and warmth in her leg was normal, despite warning the clinicians that she had a history of blood clots. Sutton-El urged her to visit the emergency room. Tests ...Read more

Ask the Pediatrician: What should I do if my child's ADHD medication is out of stock during the shortage?
If you are scrambling to get your child's attention-deficit/hyperactivity disorder prescription refilled, you are definitely not alone. Families across the U.S. have been dealing with an ADHD medication shortage first reported in October 2022 that is now well into its second year.
I've heard stories of parents and caregivers having to drive as ...Read more

Dietary choices are linked to higher rates of preeclampsia among Latinas
For pregnant Latinas, food choices could reduce the risk of preeclampsia, a dangerous type of high blood pressure, and a diet based on cultural food preferences, rather than on U.S. government benchmarks, is more likely to help ward off the illness, a new study shows.
Researchers at the USC Keck School of Medicine found that a combination of ...Read more

Did racism kill Jackie Robinson?
Baseball great Jackie Robinson was a living, breathing example of athleticism and apparent good health, playing four sports at UCLA and becoming the first Black man to play in major league baseball.
And yet, the athletic hero and civil rights champion died at age 53, almost blind, from a heart attack, with underlying diabetes and ...Read more

Rising complaints of unauthorized Obamacare plan-switching and sign-ups trigger concern
Federal and state regulators aren’t doing enough to stop the growing problem of rogue health insurance brokers making unauthorized policy switches for Affordable Care Act policyholders, say consumers, agents, nonprofit enrollee assistance groups, and other insurance experts.
“We think it’s urgent and it requires a lot more attention and ...Read more

Fact check: Biden is right about $35 insulin cap but exaggerates prior costs for Medicare enrollees
Insulin for Medicare beneficiaries “was costing 400 bucks a month on average. It now costs $35 a month.”
President Joe Biden, in a March 22 speech
____
The cost of insulin in the United States has risen considerably in recent years, with some estimates finding that Americans have paid around 10 times as much for the drug as people in ...Read more

Gov. Kelly vetoes Kansas ban on gender transition surgery, hormone therapy for trans youth
Kansas Gov. Laura Kelly on Friday vetoed a bill that would ban transgender minors from receiving gender transition surgeries and hormone therapy, setting up another veto override fight over efforts to regulate the lives of trans residents.
Kelly, a Democrat, described the legislation passed by the Republican-controlled Legislature as divisive, ...Read more

Nearly 1 in 4 adults dumped from Medicaid are now uninsured, survey finds
Nearly a quarter of adults disenrolled from Medicaid in the past year say they are now uninsured, according to a survey released Friday that details how tens of millions of Americans struggled to retain coverage in the government insurance program for low-income people after pandemic-era protections began expiring last spring.
The first ...Read more

After uphill battle, company is poised for takeover of bankrupt California hospital
MODESTO, Calif. — When American Advanced Management made a bid for the bankrupt Madera Community Hospital last year, many local officials and others involved in trying to reopen the facility didn’t take the company seriously.
The 11-year-old firm, based in Modesto, was already running a handful of small, rural hospitals, but Madera had far ...Read more
Popular Stories
- Why some adults may need another dose of measles vaccine
- More kids are dying of drug overdoses. Could pediatricians do more to help?
- Doctors take on dental duties to reach low-income and uninsured patients
- Mayo Clinic virologist offers perspective on avian influenza outbreak
- Colorado has lost dozens of autism clinics as state struggles to shore up funding
Comics





