Business
/ArcaMax

Sesame Street writers vote to strike if a new deal isn't reached Friday
Writers on the long-running kids’ show “Sesame Street” voted to strike if a new contract isn’t reached by Friday.
The Writers Guild of America East (WGAE) said 35 writers on the program “unanimously, with 100% participation” agreed to walk off the job should management fail to agree to a new collective bargaining agreement that’s ...Read more

Netflix adds 9.33 million customers, crushing Street forecasts
Netflix Inc. posted its best start to the year since 2020, attracting more new customers than anyone expected thanks to a strong slate of original programs and a crackdown on password sharing.
Netflix added 9.33 million customers in the first quarter of 2024, according to a statement Thursday, nearly doubling the 4.84 million average of ...Read more

Uber rolls out blue checkmark system for rider verification in 12 cities
Uber launched a pilot program Thursday in 12 cities around the U.S. to verify riders on the app for increased safety for drivers.
The new safety feature means riders using Uber will be verified on the app and have a blue checkmark badge added to their profile for drivers to see, according to a news release. Most accounts will be verified ...Read more

Disney, Universal report leg injuries, loss of consciousness on rides
Visitors at Walt Disney World and Universal Orlando experienced leg injuries, shortness of breath and loss of consciousness on rides at their theme parks in the first three months of 2024, a state report revealed Thursday.
The document listed eight guest injuries at Disney and two at Universal from January through March.
In January, a 63-year-...Read more
AG of Michigan to investigate $20M grant used by businesswoman to buy $4,500 coffee maker
LANSING, Mich. — Attorney General Dana Nessel's office has opened an investigation into a $20 million grant awarded to a Metro Detroit businesswoman that was used on expenses that included a $4,500 coffee maker, an $11,000 first-class plane ticket and $408,000 in salaries for two people over three months.
Nessel's office confirmed Wednesday ...Read more

Google fires 28 employees who protested Israel cloud contract
Google has terminated 28 employees after dozens of workers participated in sit-ins inside company offices earlier this week to protest the tech giant's work in Israel amid the war against Hamas in Gaza.
The protests, organized by the No Tech for Apartheid campaign, raised concerns about Google and Amazon's $1.2-billion cloud computing ...Read more

California in a jam after borrowing billions to pay unemployment benefits
California's massive budget deficit, coupled with the state's relatively high level of joblessness, has become a major barrier to reducing the billions of dollars of debt it has incurred to pay unemployment benefits.
The surge in unemployment brought on by the COVID-19 pandemic pushed the state's unemployment insurance trust into insolvency. ...Read more

Mercedes-Benz workers to vote on whether to join the UAW in May
Employees at Mercedes-Benz Group's assembly and battery plants outside Tuscaloosa, Alabama, will vote whether to join the United Auto Workers from May 13-17, the National Labor Relations Board said Thursday.
The election will be the Detroit-based union's second at a foreign-owned assembly plant following the launch of its $40 million organizing...Read more

Red state coal towns still power the West Coast. We can't just let them die
COLSTRIP, Montana — In the early morning light, it’s easy to mistake the towering gray mounds for an odd-looking mountain range — pale and dull and devoid of life, some pine trees and shrublands in the foreground with lazy blue skies extending up beyond the peaks.
But the mounds aren’t mountains.
They’re enormous piles of dirt, torn...Read more

Downtown LA is hurting. Frank Gehry thinks arts can lead a revival
With two major expansions of downtown Los Angeles cultural institutions in the works, Bunker Hill is primed to elevate its status as the region's leading arts center even as the area around it struggles with persistent homelessness and post-pandemic losses of office tenants.
Bunker Hill will soon have the largest concentration of buildings ...Read more
Real estate Q&A: How can I get my HOA to allow car covers?
Q: I recently moved and am now, for the first time, dealing with a homeowners association. I have a classic Cadillac that I used to keep covered at my previous residence. The rules in place at my new residence forbid car covers. Other nearby communities seem to allow covered cars, but not mine, so this rule was established by my association only...Read more

Media groups urge feds to investigate after Google limits California news in search results
Two journalism trade organizations representing thousands of publishers this week called on California Attorney General Rob Bonta, the U.S. Department of Justice and the Federal Trade Commission to investigate Google, after the tech giant announced Friday that it was pulling California news articles from its search platforms for some users.
...Read more

Michael Hiltzik: With his Truth Social stock, Trump may be laughing all the way to the bank -- but his investors have reason to weep
With their life savings, childrens' college funds and their own retirement prospects at stake, most people probably view investing in stocks as a serious business. Now and then, however, the markets produce comedy gold.
Hello, Trump Media & Technology Group.
The owner of Truth Social, a social media platform exclusively hitched to Donald Trump...Read more
Construction paused at VinFast's NC site as carmaker seeks a smaller footprint
Nearly nine months after VinFast broke ground on its planned $4 billion electric vehicle factory in North Carolina, construction at the Chatham County site has stalled while local officials await updated building plans from the Vietnamese carmaker.
Chatham County confirmed Tuesday what News & Observer drone footage makes clear: No significant ...Read more

Boeing workers still scared to raise safety concerns, say FAA-appointed experts
After spending a year interviewing Boeing employees and executives, documenting the company’s safety processes and taking stock of changes Boeing made after two deadly 737 Max crashes, an expert panel determined the company has much more work to do.
Among its many findings, the panel concluded Boeing employees fear retaliation if they speak ...Read more

Boeing hid safety risks in 'criminal cover-up,' whistleblowers tell Senate
In sworn testimony before a U.S. Senate hearing Wednesday, a Boeing engineer reiterated his accusation that Boeing has hidden safety risks on the 787 Dreamliner and the 777 widebody jets, rejecting the account Boeing provided Monday in an effort to reassure the public.
And a former Boeing manager accused the company of a “criminal cover-up”...Read more
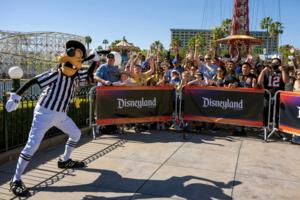
Disneyland costumed staffers from Goofy to Elsa seek union vote
Walt Disney Co. employees who dress up as Goofy and other beloved characters to entertain guests at the company’s California theme parks have taken a key step toward unionizing.
A majority of the staff members who meet with fans or participate in parades at the Disneyland Resort have signed a petition indicating they want to join the Actors�...Read more

As other banks report decline in deposits, US Bancorp experiencing a surge
As other banks report declines to their average deposits, Minneapolis-based U.S. Bancorp, the parent company of U.S. Bank National Association, has experienced a surge in that segment despite an uncertain rate environment.
For the bank's first quarter ended March 31, average total deposits increased $279 million, or 0.1%, from the fourth ...Read more

Tech layoffs jolt Bay Area economy with hundreds of new job cuts
SUNNYVALE, California — A high-profile aerospace and defense contractor and a semiconductor company were among the latest tech firms to chop jobs in the Bay Area, cutbacks that will erase more than 200 positions.
Lockheed Martin, Coherent, and Hinge Health, a tech company that provides online physical therapy, have decided to slash employment...Read more

Canada to start taxing tech giants in 2024 despite US complaints
Canada will start applying a proposed tax on the world’s biggest technology companies this year, despite threats from American lawmakers to carry out trade reprisals against a levy that will primarily hit U.S. firms.
Legislation to enact the digital services tax is currently before Canada’s Parliament. Once it passes, “the tax would begin...Read more
Popular Stories
- Michael Hiltzik: With his Truth Social stock, Trump may be laughing all the way to the bank -- but his investors have reason to weep
- False offers of cash subsidies used to 'capture' health insurance customers, lawsuit alleges
- Boeing workers still scared to raise safety concerns, say FAA-appointed experts
- Construction paused at VinFast's NC site as carmaker seeks a smaller footprint
- Red state coal towns still power the West Coast. We can't just let them die
Comics





